当前时间:2025-06-04
本文基于Shuzhen Zou等学者于2023年发表在《Frontiers in Environmental Science》的论文“Grazing disturbance increased the mobility, pathogenicity and host microbial species of antibiotic resistance genes, and multidrug resistance genes posed the highest risk in the habitats of wild animals”(下称“参考论文”),旨在为生物信息学初学者及相关领域研究人员提供一份详尽的方法学解析和深入研究论文的撰写指南。我们将聚焦于论文中的抗生素抗性基因(ARGs)检测与分析、微生物群落分析以及数据分析策略,力求内容既满足实验复现的细节需求,也兼顾方法原理的概念性理解。
研究背景简述: 全球范围内,抗生素的广泛使用加剧了抗生素抗性基因(ARGs)在环境中的传播和积累,对生态系统和人类健康构成潜在威胁。野生动物栖息地作为相对独特的生态系统,其土壤微生物群落中ARGs的状况及人类活动(如放牧)对其影响的研究日益受到关注。参考论文指出,牲畜与野生动物之间频繁发生抗生素抗性细菌及其ARGs的交换,而放牧干扰(Grazing disturbance, GD)是中国自然保护区内引起环境剧变的主要因素之一。因此,研究放牧干扰下中国自然保护区ARGs的风险,对于评估野生动物栖息地健康具有重要意义。
核心问题提炼: 本文旨在深度解析参考论文的研究方法,特别是其如何通过对比放牧干扰(GD)区与无放牧干扰的对照(CK)区土壤样品,揭示放牧活动对ARGs多样性、丰度、可移动性(mobility)、宿主微生物种类(host microbial species)以及潜在致病性(pathogenicity)的影响。参考论文进一步探讨了多重耐药基因(Multidrug resistance genes, MRGs)在野生动物栖息地中构成的最高风险,以及放牧干扰如何加剧这一风险。
分析主线: 围绕参考论文的核心技术手段,包括样品采集与处理、DNA提取、高通量测序(16S rRNA扩增子测序与宏基因组测序)、生物信息学分析(ARGs鉴定与定量、微生物群落结构分析、ARGs可移动性与致病性评估)以及统计学分析,进行方法学详解和概念性阐释。旨在帮助读者理解研究思路、掌握关键技术,并为开展同类型研究提供参考。
方法学详解 (实验复现导向):
研究地点: 中国四川省白河国家级自然保护区。该保护区是濒危野生动物如大熊猫 (Ailuropoda melanoleuca) 和川金丝猴 (Rhinopithecus roxellana) 的重要栖息地。然而,该区域未被纳入中国大熊猫国家公园体系,存在管理相对宽松的问题。当地原住民依赖畜牧业为生,其畜牧方式较为粗放,放牧痕迹甚至与川金丝猴的活动范围重叠 (Liu et al., 2021; Yuan, 2018)。
实验分组:
采样策略:
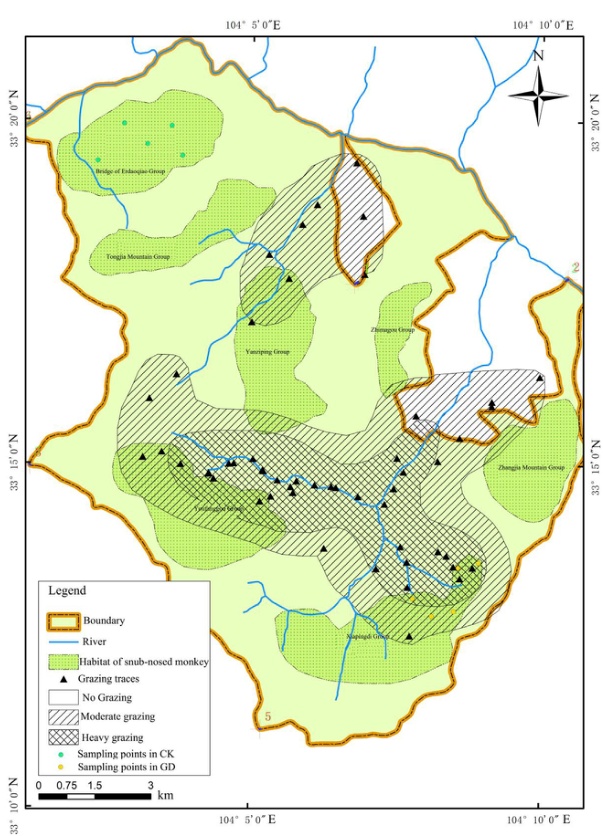
图1. 白河国家级自然保护区及采样点示意图 (引用自参考论文中的图1)。图中清晰标示了放牧干扰区(GD)和对照区(CK)的采样点位置。来源:Zou et al., 2023
概念性理解 (初学者导向):
方法原理: 对照研究是科学研究中的基本方法。通过选择一个受特定因素(此处为放牧)干扰的实验组(GD)和一个不受该因素干扰的对照组(CK),并在其他条件尽可能一致的情况下进行比较,可以较为可靠地推断该因素所产生的影响。采样点选择的代表性和采样的随机性、重复性是保证结果普适性和统计学效度的关键。
应用场景: 此方法广泛应用于生态学、环境科学等领域,用于评估人类活动(如农业开垦、工业污染、旅游开发、放牧)对自然生态系统(如土壤、水体、森林)的物理、化学及生物学特性的影响。
要点提示/注意事项:
方法学详解 (实验复现导向):
DNA提取试剂盒: FastDNA® SPIN Kit for Soil (MP Biomedicals, United States),货号及具体批次未在论文中说明,但可通过MP Biomedicals官网查询。
起始样品量: 每个DNA提取使用2克冷冻土壤。
操作流程: 严格按照FastDNA® SPIN Kit for Soil试剂盒的制造商操作手册进行。该试剂盒通常包括机械破碎(配合裂解基质珠)和化学裂解步骤,然后通过硅胶膜离心柱纯化DNA。
样品合并策略: 从每个大样方中采集的三个土壤样品分别提取DNA后,将这三个DNA提取液等量混合,形成代表该大样方的一个DNA样品,用于后续的测序分析。这意味着,如果GD和CK组各有5个大样方,则最终各有5个混合DNA样品用于测序。
DNA质量检测:
仪器: NanoDrop 2000分光光度计 (Thermo Fisher Scientific, Wilmington, DE, United States)。
检测指标:
DNA浓度 (ng/µL): 评估DNA产量是否足够后续实验。
DNA纯度: 通过吸光度比值评估。A260/A280比值理想范围为1.8-2.0,低于1.8可能表示蛋白质污染,高于2.0可能表示RNA污染。A260/A230比值理想范围通常大于1.8(甚至>2.0),低于此值通常表示存在腐殖酸、酚类或碳水化合物等有机杂质污染。
概念性理解 (初学者导向):
方法原理:土壤总DNA提取旨在从复杂的土壤基质中分离出所有微生物(细菌、真菌、古菌等)的基因组DNA。FastDNA® SPIN Kit for Soil这类试剂盒通常结合物理方法(如研磨珠震荡破碎细胞)和化学方法(如裂解液中的去污剂、酶)来高效裂解微生物细胞,释放DNA。随后,利用DNA在特定化学条件下(如高盐、低pH)与硅胶膜的特异性吸附能力,将DNA吸附到离心柱的硅胶膜上,通过洗涤步骤去除蛋白质、RNA、腐殖酸等杂质,最后用低盐缓冲液或水将纯净的DNA洗脱下来。
应用场景: 从土壤、沉积物、粪便等富含抑制物的复杂环境样品中提取高质量微生物总DNA,广泛应用于微生物多样性研究(如16S rRNA测序、ITS测序)、宏基因组学、宏转录组学等研究领域。
要点提示/注意事项:
方法学详解 (实验复现导向):
目标基因与高变区: 细菌16S rRNA基因的V3-V4高变区。这是一个广泛用于细菌多样性研究的区域,兼具一定的保守性和足够的变异性以区分不同分类单元。
PCR扩增引物:
PCR扩增条件: 参考论文中未提供详细的PCR反应体系和热循环程序。通常,一个典型的PCR体系包含:DNA模板 (10-50 ng),正反向引物 (各0.2-1 µM),dNTPs (各200 µM),高性能DNA聚合酶 (如 KAPA HiFi HotStart ReadyMix, Q5 High-Fidelity DNA Polymerase),相应的PCR缓冲液,以及无核酸酶水补足体系。热循环程序一般包括:初始变性 (如95-98°C, 2-5 min),若干个循环的变性 (95-98°C, 15-30 s)、退火 (50-60°C, 15-30 s)、延伸 (72°C, 30-60 s),以及最终延伸 (72°C, 5-10 min)。具体退火温度和延伸时间需根据引物Tm值和目标片段长度优化。
文库构建与测序:
原始数据存储: 数据已提交至NCBI Sequence Read Archive (SRA) 数据库,登录号为PRJNA896787。
16S rRNA数据预处理与OTU/sOTU分析:
sOTU (sub-Operational Taxonomic Unit) 生成: 参考论文中提及使用QIIME2 (Bolyen et al., 2019) (具体版本未指明,但引用了Xia et al., 2022) 进行sOTU分析。sOTU通常指代ASVs (Amplicon Sequence Variants),它们是通过DADA2或Deblur等算法获得的精确到单核苷酸差异的序列变体,能提供比传统OTU聚类(如97%相似度)更精细的物种分辨。
测序深度标准化 (Normalization): 为了在不同样本间公平比较sOTU的丰度,将每个样本的测序深度(reads数量)均一化至41,723条reads。这可以通过抽样(rarefaction)或比例转换等方法实现。
物种分类注释 (Taxonomic assignment): 使用QIIME2的feature-classifier插件,基于Greengenes数据库 (v. 13_8, DeSantis et al., 2006),以97%的序列相似性阈值对sOTUs进行物种分类注释,获得每个sOTU从界到属(甚至种,如果数据库和序列质量允许)的分类信息。
概念性理解 (初学者导向):
方法原理: 16S rRNA基因是原核生物核糖体小亚基的组成部分,几乎所有细菌都拥有该基因。它由保守区和高变区(V1-V9)交替排列组成。保守区序列差异小,适合设计通用引物;高变区序列差异大,可用于区分不同细菌种类。通过PCR扩增特定高变区(如V3-V4),然后进行高通量测序,可以获得样品中大量细菌的16S rRNA序列片段。通过生物信息学分析这些序列,可以推断样品中细菌群落的组成结构和多样性。
应用场景: 广泛应用于环境(土壤、水体、空气)、宿主相关(肠道、口腔、皮肤)等各种样本的细菌群落结构研究,是微生物生态学研究的基石技术之一。
核心概念释义:
要点提示/数据解读初步:
方法学详解 (实验复现导向):
文库构建:
测序平台与策略:
原始数据存储: 数据已提交至NCBI SRA数据库,登录号为PRJNA906922。
宏基因组数据预处理、组装与基因预测:
质量控制 (Quality control): 具体方法参考了 Chen et al. (2021)。通常包括:
使用FastQC (Andrews, 2010) 评估原始reads质量。
使用Trimmomatic (Bolger et al., 2014) 或Cutadapt (Martin, 2011) 去除测序接头 (adapters)、低质量碱基 (通常基于Phred quality score,如Q<20或Q<30的碱基)、短读长序列 (如长度<50 bp的reads)。
(可选) 去除宿主污染:如果样本来源于宿主相关环境,可能需要比对到宿主基因组以去除宿主来源的reads。对于土壤样本,此步骤通常不必要,除非有特定植物或大型动物DNA污染的担忧。
论文中提及10个土壤样品共产生3,844,430条reads (原文为384443,此处按逻辑修正,但需以原文为准,若原文数字准确则数据量过少)。【**再次强调**:原文数字为384,443 reads for 10 samples,平均每个样本约38k reads。这对于宏基因组分析(尤其是有组装需求的)是非常低的数据量。在实验复现时,应确保足够的测序深度,例如每个样本至少10-20 Gbp的clean data。】
序列组装 (Assembly):
工具: IDBA-UD (Iterative De Bruijn Graph Assembler for Uneven Depth sequencing data, Peng et al., 2012),这是一版适合宏基因组这种测序深度不均一数据的组装软件。具体参数设置参考 Chen et al. (2021),通常包括k-mer范围的设定。
筛选标准: 保留组装后长度大于500 bp的Contigs (连续序列)。
组装结果: 从10个宏基因组样本中共获得7,635,863条contigs,平均长度为828.42 bp。
基因预测 (Gene Prediction / ORF calling):
工具: Prodigal (Prokaryotic Dynamic Programming Genefinding Algorithm, Hyatt et al., 2010) v2.6.3版本,使用-p meta参数(宏基因组模式)。
预测对象: 在组装得到的Contigs上预测开放阅读框 (Open Reading Frames, ORFs),即潜在的蛋白质编码基因。
基因覆盖度计算 (Contig/Gene Coverage):
工具: bbmap (Bushnell B. - BBMap)。
方法: 将质控后的clean reads回比对 (map back) 到组装好的contigs上。
目的: 计算每个contig或ORF被reads覆盖的程度,这可以作为其在样本中相对丰度的一个指标,也有助于评估组装的可靠性(低覆盖度区域组装可能不准确)。参考论文中 (Ju et al., 2019) 提及使用此步骤。
概念性理解 (初学者导向):
方法原理: 宏基因组鸟枪法测序 (Shotgun Metagenomic Sequencing) 是一种不依赖于培养、不针对特定基因的测序方法。它通过对环境样品中所有微生物的总DNA进行随机打断、构建文库并进行高通量测序,旨在获得尽可能全面的遗传信息。通过生物信息学分析,可以重构部分基因组序列 (contigs),预测基因功能,从而了解微生物群落的物种组成、功能潜力和基因多样性。
应用场景: 特别适用于研究微生物群落的功能概况,如代谢通路、抗生素抗性基因、毒力因子等。同时也能用于物种组成分析(通常比16S rRNA测序更准确,能鉴定到种甚至株水平,并覆盖病毒、真菌等),以及发现新的基因资源和微生物物种。
核心概念释义:
-p meta) 经过优化,适合处理片段化和测序深度不均的宏基因组数据。要点提示/数据解读初步:
方法学详解 (实验复现导向):
ARGs鉴定流程:
将上一步宏基因组分析中预测出的所有ORFs蛋白序列作为查询序列 (query sequences)。
选择一个综合性的、高质量的ARGs参考数据库。参考论文中使用的是CARD (Comprehensive Antibiotic Resistance Database, Jia et al., 2017)。这是一个持续更新的、包含已知ARGs序列、抗性机制和相关信息的权威数据库。
使用快速序列比对工具。参考论文中使用DIAMOND (v0.7.9或更新版本, Buchfink et al., 2015),它在保持较高灵敏度的同时比BLASTP快得多,适合大规模宏基因组数据的比对。
设置严格的比对和筛选阈值:
ARGs丰度计算与标准化:
计算目标: 评估每个鉴定出的ARG-like ORF在样本中的相对丰度。
计算公式 (参考论文公式1):
Relative abundance (copies/Gb) = Σ (N × read_length / L) / G_dataset
其中:
n: 一个样本中ARG-like ORFs的总数 (公式中的 Σ 代表对样本中所有ARG-like ORFs进行求和,但通常是计算单个ARG的丰度然后汇总或按类别汇总)。论文公式描述 "where n is the total number of ARG-like ORFs in one sample",这可能指对一个样本中所有ARG的总体丰度的计算,但更常见的做法是计算每个ARG的丰度。如果按单个ARG计算,则是 Relative abundance_i = (N_i × read_length / L_i) / G_dataset。这里以论文提供的汇总公式为准进行解释,但实际分析中常按单个ARG计算。
N: 比对到(覆盖)某个特定ARG-like ORF的clean reads的数量。通过将质控后的宏基因组reads回比到这个ORF序列上获得。
read_length: Clean reads的平均长度 (bp)。对于PE150测序,通常假设拼接/打断后的片段进行比对,此处论文中直接用150 bp代入,可能指参与比对的reads的长度。
L: 目标ARG-like ORF的长度 (bp)。
G_dataset: 该样本的clean reads总数据量大小,单位为Gb (Gigabase pairs)。
公式解读: (N × read_length / L) 这一项可以理解为:如果一个ORF全长被reads覆盖,且reads长度恰好等于ORF长度,则覆盖这条ORF的reads数目就是N。如果reads比ORF短,则需要 N × read_length / L 条reads才能完全覆盖L长度的ORF一次。因此,该项估算了这条ORF被测序数据覆盖的“拷贝数”或“深度归一化后的计数”。然后除以整个数据集的大小G,得到每Gb数据的拷贝数,实现了跨样本的标准化。这一方法参考了 Ma et al. (2015) 和 Xiong et al. (2018)。
丰度单位: "copies/Gb" 或 "ppm" (parts per million of reads,如果用reads数标准化)。参考论文的单位是 "copies/Gb"。
概念性理解 (初学者导向):
分析流程概述: 简而言之,就是将我们从土壤DNA中找到的所有可能的基因(ORFs),去和一个巨大的已知“坏基因”(ARGs)黑名单(CARD数据库)做比对。如果某个基因和黑名单上的某个坏基因长得很像(满足E-value和Identity阈值),我们就把它标记为“疑似坏基因”(ARG-like ORF)。然后,我们数一数每个样品里这种“疑似坏基因”有多少条,再根据每个样品总共测了多少数据进行标准化,这样就可以公平地比较不同样品中ARGs的多少了。
核心概念释义:
结果解读要点: 通过ARGs的鉴定,可以知道样品中存在哪些类型的抗性基因(如耐四环素的、耐β-内酰胺的等)。通过丰度计算和比较,可以了解不同处理组(如放牧区vs对照区)中ARGs的总体水平以及特定类型ARGs的富集情况。
方法学详解 (实验复现导向):
移动遗传元件 (Mobile Genetic Elements, MGEs) 的鉴定:
参考数据库: 使用MGE数据库,参考论文中链接指向了Aclame database (http\://aclame.ulb.ac.be/, Leplae et al., 2010)。Aclame数据库收集了噬菌体、质粒和原噬菌体等可移动遗传元件的信息。其他常用的MGE数据库还包括ISfinder (针对插入序列, Siguier et al., 2006), INTEGRALL (针对整合子, Moura et al., 2009) 等。实际操作中可能需要整合多个MGE数据库或使用更全面的MGE识别工具。
比对工具与参数: 使用BLASTP (蛋白序列比对蛋白数据库) 将宏基因组预测的ORFs与MGE数据库中的蛋白序列进行比对。
E-value: ≤ 10⁻⁵。
鉴定标准: 比对结果中共享序列一致性 (Shared Identity) ≥ 80% 的ORFs被鉴定为MGEs。
MGEs丰度标准化: 同ARGs的丰度计算方法,即标准化为 "copies/Gb"。
ARGs与MGEs的关联分析 (评估ARGs的可移动性):
判断标准: 如果一个ORF上同时注释到了ARG (根据3.1节的方法) 和MGE (根据本节上述方法),则认为该ARG可能通过此MGE进行移动。这一逻辑源自 Ju et al. (2019),他们认为MGE与ARG在同一个组装的contig上或同一个预测的ORF上,表明两者可能存在物理连接,从而ARG具有被该MGE介导转移的潜力。
具体分析方法 (推测,论文未详述):
概念性理解 (初学者导向):
方法原理: 抗生素抗性基因的横向基因转移 (Horizontal Gene Transfer, HGT) 是ARGs在细菌种内和种间快速传播和扩散的主要途径,对公共卫生构成严重威胁。MGEs是HGT的主要载体,它们如同“基因出租车”,能够携带包括ARGs在内的各种基因在不同细菌之间穿梭。通过分析ARGs与MGEs在基因组上的物理关联(如同一个ORF上,或在同一个contig上靠得很近),可以评估ARGs通过HGT传播的潜力。
应用场景: 评估环境中ARGs的传播风险,识别高风险ARGs(即易于转移的ARGs),理解抗性基因扩散的分子机制,为制定阻断ARGs传播的策略提供依据。
核心概念释义:
要点提示/注意事项:
方法学详解 (实验复现导向):
毒力因子 (Virulence Factors, VFs) 的鉴定:
参考数据库: VFDB (Virulence Factor Database, Liu et al., 2022)。VFDB是一个全面的、人工整理的细菌毒力因子数据库,包含已知VFs的序列、功能、来源菌株等信息。参考论文中使用的是VFDB的setB (a set of core dataset representing proteins with experimentally verified VFs)。
分析工具/比对方法: 使用VFanalyzer工具套件中的方法(通常是基于BLAST的比对)将宏基因组预测的ORFs与VFDB数据库进行比对。参考论文中未明确提及VFanalyzer,但其逻辑与VFDB的使用是一致的,可能是通过DIAMOND或BLASTP比对到VFDB。
比对与筛选参数:
E-value: ≤ 10⁻¹⁰。
鉴定标准: 比对结果中序列一致性 (Identity) ≥ 80% 的ORFs被鉴定为潜在的毒力因子基因 (VFGs)。
耐药致病菌 (Drug-Resistant Pathogens) 的判定与风险评估:
核心判断标准 (参考 Zhang et al., 2021): 如果一个ORF同时被鉴定为ARG-like ORF (携带抗性基因) 并且也被鉴定为携带VF (毒力因子),那么这个ORF所在的细菌就被认为是潜在的耐药致病菌。这种细菌由于既能抵抗抗生素治疗,又具有致病能力,因此构成了最高的ARGs相关风险。
分析流程:
论文中的发现:
共检测到26个ARG-like ORFs同时也携带VFs,这些ORFs分属于三种致病菌:Klebsiella pneumoniae subsp. pneumoniae NTUH-K2044, Acinetobacter baumannii ACICU, 和 Neisseria meningitidis MC58。
GD组中这类“双重功能”ORFs的总数显著高于CK组。
在GD组中检测到6种仅在该组出现的“双重功能”ORFs,它们分属于Klebsiella pneumoniae NTUH-K2044 和 Acinetobacter baumannii ACICU。特别是,Acinetobacter baumannii ACICU这个耐药致病菌仅在GD组检出。
概念性理解 (初学者导向):
方法原理: 抗生素抗性基因本身并不会直接导致疾病,但当ARGs存在于能够引起疾病的细菌(即病原菌)中时,这些病原菌就变成了耐药病原菌。耐药病原菌感染宿主后,常用的抗生素治疗可能无效或效果减弱,导致治疗失败、病情迁延甚至死亡。毒力因子是病原菌致病的关键“武器”。因此,通过同时检测一个微生物是否携带ARGs和VFs,可以识别出潜在的耐药致病菌,从而评估ARGs带来的直接健康风险。
应用场景: 评估环境中ARGs对人类和动物健康的潜在威胁,尤其是在医院、养殖场、污水处理厂等可能存在病原菌与ARGs交汇的环境中。识别高风险的耐药病原菌,为制定防控措施提供科学依据。
核心概念释义:
要点提示/注意事项:
方法学详解 (实验复现导向):
sOTU聚类与物种注释: 已在2.1节中详细描述。核心步骤包括使用QIIME2流程,通过DADA2或类似算法生成sOTUs/ASVs,然后基于Greengenes 13_8数据库 (97%相似度) 进行物种分类注释。
Alpha多样性指数计算与比较:
指数类型:
Simpson's多样性指数: 该指数衡量群落中物种多样性,同时考虑物种数量和各物种的相对丰度。其计算公式通常为 D = Σ (n_i * (n_i-1)) / (N * (N-1)),其中n_i是第i个物种的个体数,N是所有物种的总个体数。常用的Simpson多样性指数是 1-D (Gini-Simpson index) 或 1/D (Inverse Simpson index),这些指数值越大,多样性越高。参考论文中提到“Simpson's index gives more weight to the more abundant species”。
ACE (Abundance-based Coverage Estimator) 多样性指数: 基于丰度的覆盖度估计量,用于估算群落中的物种丰富度,特别是对稀有种的检测较为敏感。ACE指数值越大,物种丰富度越高。
计算工具: 可使用QIIME2内置的qiime diversity alpha命令,或者在R中使用vegan包 (Oksanen et al., 2020) 中的diversity()函数(计算Shannon, Simpson)和estimateR()函数(计算ACE, Chao1)。
统计比较: 使用t-检验比较GD组和CK组之间各Alpha多样性指数的均值差异,p < 0.05认为差异显著。
参考论文发现: GD组土壤微生物的Simpson多样性显著高于CK组 (p = 0.032),而ACE多样性在GD组显著低于CK组 (p = 0.012)。这表明放牧干扰降低了土壤微生物的总物种数 (以ACE衡量),但增加了群落中优势微生物种类的优势度 (Simpson指数对优势种敏感,其升高可能反映优势种更突出,或物种分布更不均匀但优势种相对丰度组合导致指数升高)。【注:关于Simpson指数的解读,高值通常代表高多样性,但其对优势种的权重较大。如果优势种变得更“优势”,其他物种变得更稀有,总物种数减少,Simpson指数(1-D)可能会升高,也可能降低,取决于具体计算的Simpson指数形式以及物种分布变化的具体情况。作者的解释是GD增加了优势种的优势度,导致Simpson多样性增加。】
Beta多样性分析 (群落结构差异):
距离矩阵计算: 虽然论文方法部分未明确指出所用的距离算法,但讨论中提到了群落结构。常用的距离测度包括:
Bray-Curtis相异度: 基于物种丰度数据,不考虑物种有无,对丰度变化敏感。
Jaccard相异度: 基于物种有无(presence/absence)数据,不考虑丰度。
Unweighted UniFrac距离: 基于物种有无数据,并考虑物种间的系统发育关系。
Weighted UniFrac距离: 基于物种丰度数据,并考虑物种间的系统发育关系。
计算通常在QIIME2 (qiime diversity beta 或 qiime diversity beta-phylogenetic) 或R (vegan::vegdist, phyloseq::distance) 中完成。
排序分析与可视化:
PCoA (Principal Coordinate Analysis, 主坐标分析): 基于距离矩阵进行降维,将高维的群落数据在二维或三维空间中展示样本间的相似性/差异性。
NMDS (Non-metric Multidimensional Scaling, 非度量多维尺度分析): 也是一种基于距离矩阵的降DIY方法,它不依赖于数据的线性假设,通常能更好地反映样本间的真实距离关系,但计算压力较大。
可视化通常在R中使用ggplot2 (Wickham, 2016) 结合vegan或phyloseq (McMurdie & Holmes, 2013) 包完成。
组间差异显著性检验:
PERMANOVA (Permutational Multivariate Analysis of Variance): 基于距离矩阵的非参数多元方差分析,用于检验不同分组(如GD vs CK)之间微生物群落结构的质心是否存在显著差异。在R中通过vegan::adonis2实现。
ANOSIM (Analysis of Similarities): 另一种基于距离矩阵的非参数检验方法,比较组内距离与组间距离的差异。
优势菌群与差异菌群分析:
图2. 模拟放牧干扰(GD)与对照(CK)组土壤微生物Alpha多样性指数比较 (t-test, * p < 0.05)。数据为示意,基于参考论文结论:GD组Simpson指数显著高于CK,ACE指数显著低于CK。
概念性理解 (初学者导向):
核心概念释义:
Alpha多样性 (α-diversity): 指单个样本或特定生境内的物种多样性。它包含两个主要方面:
物种丰富度 (Species Richness): 群落中物种的数量。如Observed OTUs/ASVs, Chao1指数, ACE指数。
物种均匀度 (Species Evenness): 群落中各物种个体数量分布的均匀程度。Pielou's evenness指数是常用指标。
综合指数 (如Shannon, Simpson): 同时考虑物种丰富度和均匀度的多样性指数。
Beta多样性 (β-diversity): 指不同样本或不同生境之间的物种组成差异程度,也称为生境间多样性。通过计算样本间的距离或相异度来衡量。
UniFrac距离: 一种特殊的beta多样性距离度量,它整合了物种的系统发育信息。Unweighted Unifrac只考虑物种有无和进化树,Weighted Unifrac则同时考虑物种丰度和进化树。
指数解读与应用:
要点提示:
方法学详解 (实验复现导向):
鉴定对象: 携带ARGs的开放阅读框 (ORFs),这些ORFs是在宏基因组组装的contigs上预测得到的。
比对数据库与工具:
物种注释与宿主判定:
分析重点与论文发现:
概念性理解 (初学者导向):
方法原理: 确定哪些微生物携带ARGs是理解ARGs在环境中传播和风险的关键。通过宏基因组测序获得了包含ARGs的基因片段(ORFs on contigs)。将这些ORF序列与大型的、经过注释的参考基因组数据库(如NCBI RefSeq)进行比对,可以找到这些ORF最可能来源于哪些微生物。MEGAN等工具利用LCA算法,综合多个比对结果来给ORF一个最可靠的物种归属。如果一个携带ARG的ORF被鉴定出其宿主菌,就明确了该ARG在该微生物体内的存在。
应用场景: 追踪ARGs在微生物群落中的分布,识别携带ARGs的关键微生物类群(尤其是病原菌或与人类/动物健康密切相关的细菌),分析不同环境条件下ARGs宿主谱的变化。
核心概念释义:
要点提示/注意事项:
方法学详解 (实验复现导向):
数据准备:
相关性计算:
vegan包提供了计算Spearman相关系数的功能 (如cor()函数配合method="spearman",或专门的生态学相关性函数)。网络构建与可视化:
构建标准 (边形成的条件):
相关系数 (ρ, rho): |ρ| ≥ 0.8。即Spearman相关系数的绝对值大于等于0.8,表明sOTU丰度与ARG亚型丰度之间存在强相关关系(正或负)。
显著性 (p-value): p ≤ 0.01。即相关性检验的p值小于等于0.01,表明观察到的强相关性是统计学显著的,而非随机产生的。
网络构建工具: R语言中的igraph包 (Csardi & Nepusz, 2006) 是一个强大的网络分析和可视化工具,可以基于相关性矩阵构建网络对象。
网络可视化工具: Gephi (version 0.9.5, Bastian et al., 2009) 是一款开源的交互式网络可视化和探索平台,可以将igraph等生成的网络数据导入进行美化和分析。
网络拓扑参数分析与解读:
节点 (Nodes): 网络中的sOTUs和ARG亚型。
边 (Edges): 连接节点之间的显著相关关系(红线表示正相关,绿线或其他颜色表示负相关)。
分析重点: 比较GD组和CK组网络的复杂性,如节点数、边数、连接度 (degree)、模块化 (modularity)、关键节点 (keystone species/ARGs) 等。
参考论文发现:
CK组网络: 271个节点,520条边 (56条负相关, 462条正相关)。
GD组网络: 305个节点,1150条边 (60条负相关, 1090条正相关)。
结论: GD组的微生物群落与ARGs之间的关联网络更为复杂(节点和边更多),表明放牧干扰可能增强了微生物与ARGs之间的互作,或使得ARGs的分布与更多微生物类群产生关联。这间接反映了GD增加了微生物携带和转移ARGs的潜力。
图3. 模拟微生物-ARGs共现网络示意图。节点代表sOTUs (圆形) 或ARG亚型 (方形),颜色可代表不同组别或丰度。边的颜色代表正/负相关,粗细代表相关性强度。此图为通用示意,实际分析需用真实数据生成。
概念性理解 (初学者导向):
方法原理: 共现网络分析是基于“生态位重叠”或“相互作用”的假设:如果两种生物(或基因)在多个不同环境样本中的丰度变化趋势高度一致(同增同减,即强正相关)或完全相反(一增一减,即强负相关),则它们之间可能存在某种生态学上的关联。这种关联可能是直接的相互作用(如捕食、共生、竞争),也可能是对同一环境因素的相似或相反响应,或者是宿主-基因关系(如特定细菌携带特定ARG)。通过构建网络,可以将这些复杂的关联关系可视化,并识别出网络中的关键“角色”。
应用场景: 探索微生物群落内部物种间的相互作用;识别对群落结构或功能起关键作用的物种(关键种);分析基因(如ARGs)与特定微生物类群的潜在宿主关系或共现模式;比较不同环境条件下微生物网络的结构和稳定性差异。
核心概念释义:
Spearman相关性: 一种非参数统计方法,衡量的是两个变量单调相关的程度。它不对数据的分布做特定假设,因此广泛用于处理非正态的生态学数据(如物种丰度)。
共现网络 (Co-occurrence Network):
节点 (Nodes/Vertices): 代表研究的实体,如本研究中的sOTUs和ARG亚型。
边 (Edges/Links): 代表节点之间的显著相关关系。边的权重可以表示相关性的强度,边的类型可以表示正负相关。
网络拓扑参数: 用于描述网络结构的量化指标,如:
节点度 (Degree): 一个节点连接的边的数量,反映节点在网络中的连接中心性。
连接度 (Connectivity): 与节点度类似,有时指加权度(考虑边的权重)。
模块性 (Modularity): 网络中形成紧密连接子集的程度。模块内连接紧密,模块间连接稀疏。
关键节点 (Keystone species/hubs): 在网络中具有高度中心性(如高连接度、高介数中心性)或连接不同模块的节点,它们的移除可能对网络结构和功能产生较大影响。
igraph/Gephi: igraph是R和Python中常用的网络分析包,提供网络构建、参数计算、布局算法等功能。Gephi是一款强大的交互式网络可视化软件,擅长大型网络的探索和美化。
要点提示/注意事项:
方法学详解 (实验复现导向):
核心目的: 通过统计学方法比较放牧干扰组 (GD) 和对照组 (CK) 之间各项量化指标(如多样性指数、基因丰度、特定微生物类群丰度等)的均值是否存在显著差异,从而判断放牧干扰是否对这些指标产生了实质性影响。
常用统计检验方法:
t-检验 (t-test): 参考论文中明确提及使用t-检验比较GD和CK组之间的差异 (p < 0.05为显著)。这通常指独立样本t检验,用于比较两组独立样本的均值。
前提条件: 理论上,标准t检验要求两组数据均来自正态分布总体且两组方差相等(方差齐性)。
变体: 若方差不齐,可使用Welch's t-test。若数据严重偏离正态分布且样本量较小,应考虑非参数检验。
非参数检验 (如Wilcoxon秩和检验/Mann-Whitney U检验): 当数据不满足t检验的正态性或方差齐性假定时,或者数据为等级数据时,非参数检验是更稳健的选择。它比较的是两组数据的中位数或分布是否存在差异,而非均值。虽然参考论文主要提及t检验,但在微生物组数据分析中,由于数据常呈偏态分布,Wilcoxon检验也十分常用。
方差分析 (ANOVA): 如果研究涉及两个以上的分组(例如,不同放牧强度、不同放牧时间点),则应使用ANOVA进行多组均数比较。若ANOVA结果显著,还需进行事后多重比较(post-hoc tests,如Tukey's HSD, Bonferroni, Dunnett's test等)来确定具体哪些组之间存在差异。
显著性水平 (Significance Level, α): 通常设定为0.05。当检验得到的p-value < α时,拒绝原假设(例如,两组均值相等),认为观察到的差异是统计学显著的。
多重比较校正: 当对多个指标进行独立的假设检验时,会增加犯第一类错误(假阳性)的概率。因此,需要对p值进行校正,常用的方法有Bonferroni校正(非常严格)和FDR (False Discovery Rate) 控制方法(如Benjamini-Hochberg, BH法,相对宽松且控制力好)。参考论文中未明确说明是否对所有t检验结果进行了多重比较校正,但在实践中,特别是涉及大量基因或OTU的比较时,此步骤非常重要。
数据可视化: 显著性差异通常在图表(如箱线图、柱状图)中用星号 (* p < 0.05, ** p < 0.01, *** p < 0.001) 或字母标记法表示。
软件实现: R语言是进行统计分析和可视化的强大工具,其内置函数(如t.test(), wilcox.test(), aov())和各种包(如ggplot2, ggpubr, rstatix)可以方便地实现上述检验和可视化。SPSS, GraphPad Prism等统计软件也广泛使用。
参考论文中应用t检验的指标示例:
图4. 模拟GD组与CK组多重耐药基因(MRGs)相关风险指标比较。如MRGs的Simpson多样性、携带MRGs的质粒ORFs数量、耐药病原体中MRGs相关ORFs数量。数据为示意,基于参考论文结论并假设GD组风险指标均显著高于CK组。(* p < 0.05, ** p < 0.01)
概念性理解 (初学者导向):
基本原理: 统计检验的核心思想是判断观察到的样本差异在多大程度上仅仅是由于抽样随机性造成的,还是确实反映了总体之间的真实差异。通过计算检验统计量(如t值、F值)并得到对应的p值,与预设的显著性水平α进行比较,从而做出推断。
P-value的理解: P值是在原假设(如两组均值无差异)为真的前提下,观察到当前样本结果或更极端结果的概率。P值越小,越有理由拒绝原假设。它不是原假设为真的概率,也不是实验结果可重复的概率。
效应量 (Effect Size): P值只能告诉我们差异是否“统计学上显著”,但不能告诉我们差异的“实际大小”或“生物学意义”。效应量指标(如Cohen's d, R², Odds Ratio等)可以衡量差异的幅度。在论文中同时报告P值和效应量,能提供更全面的信息。虽然参考论文未突出效应量,但在撰写高质量论文时建议考虑。
选择合适的统计方法: 正确选择统计方法是保证结论可靠性的前提。需要考虑:
对于微生物组数据,由于其非正态、稀疏、组成性等特点,经常需要使用非参数检验或专门针对此类数据开发的统计模型。
方法学详解 (实验复现导向):
参考论文并未采用单一的、标准化的定量生态风险评估模型(如基于预测无效应浓度PNEC和环境检测浓度MEC计算风险商RQ的方法),而是通过一种综合证据、多维度分析的逻辑框架来评估放牧干扰(GD)对土壤ARGs所带来的生态风险。这种评估更加侧重于ARGs的赋存特征变化、传播潜力增强以及与致病性关联的增强,是基于多个生物信息学分析结果的整合解读。其评估逻辑可概括为以下层面:
ARGs本身的特征变化是否指示风险增加?
ARGs的横向转移潜力 (HTP) 是否增强?
ARGs与致病性的关联是否增强?
宿主微生物群落的响应是否有利于ARGs的维持与传播?
对多重耐药基因 (MRGs) 风险的特别关注:
概念性理解 (初学者导向):
基于对参考论文的深度解析,为计划开展同类型研究并撰写论文的科研人员提供以下结构性建议。请注意,这仅为通用框架,具体内容需根据您的实际研究结果进行填充和调整。
引言部分旨在为研究提供背景,阐明研究的必要性和创新性,并清晰地提出研究问题和假设。
背景铺垫:
文献缺口/科学问题:
回顾现有关于人类活动对环境中ARGs影响的研究,特别是针对农业、畜牧业等活动。
指出当前研究在特定方面可能存在的不足,例如:
对自然保护区内特定人类干扰(如传统放牧)对土壤ARGs的系统性影响研究较少。
多数研究可能侧重于ARGs的丰度和多样性,而对其可移动性、致病性潜力及其与宿主微生物群落动态互作的综合评估不够深入。
针对特定地理区域(如您研究的区域)或特定野生动物栖息地的此类研究可能缺乏。
基于上述文献缺口,凝练出本研究的核心科学问题。例如:在[您的研究区域]的[特定野生动物]栖息地,传统的放牧活动是如何影响土壤微生物群落介导的ARGs的(1)赋存特征(类型、丰度、多样性)?(2)横向转移潜力(通过MGEs的传播风险)?(3)潜在致病性(与毒力因子的关联及潜在宿主)?以及(4)哪类ARGs(如MRGs)构成主要风险?
研究目的与假设:
研究意义:
材料与方法部分应提供足够详细的信息,以便其他研究者能够重复您的实验。务必清晰、准确、条理分明。
研究区域与实验设计 (Study Area and Experimental Design):
样品采集与处理 (Sample Collection and Pretreatment):
土壤理化性质测定 (Soil Physicochemical Properties Analysis): (虽然参考论文未重点分析,但通常是相关研究的重要组成部分,用以解释微生物和ARGs变化的潜在驱动因素)
土壤总DNA提取与质量控制 (Soil Total DNA Extraction and Quality Control):
高通量测序 (High-Throughput Sequencing):
16S rRNA基因扩增子测序:
目标区域与引物: 明确扩增的16S rRNA高变区(如V3-V4)及所用的通用引物序列(如338F/806R),注明引物是否带有Barcode和测序接头。引物合成公司。
PCR扩增体系: 详细列出PCR反应总体积及各组分名称和终浓度(如模板DNA量、引物浓度、dNTPs浓度、聚合酶种类和用量、Buffer成分)。
PCR热循环程序: 详细描述初始变性、循环(变性、退火、延伸的温度、时间、循环数)、最终延伸的条件。
PCR产物纯化与文库构建: 纯化方法(如磁珠回收),文库构建方法(如双Index法),文库质检(如Agilent Bioanalyzer)。
测序平台与策略: 如Illumina MiSeq平台,PE250 (2×250 bp)或PE300 (2×300 bp)双端测序。预期每个样本的有效数据量(reads数或数据大小)。测序服务公司。
宏基因组鸟枪法测序:
DNA片段化与文库构建: 起始DNA量,DNA打断方法(如超声波破碎Covaris),文库类型(如PCR-free或标准文库),目标插入片段大小(如300-500 bp),文库构建试剂盒名称和厂家。
测序平台与策略: 如Illumina NovaSeq 6000平台,PE150 (2×150 bp)双端测序。预期每个样本的数据产出(以Gbp为单位)。测序服务公司。
数据提交: 将原始测序数据提交到公共数据库(如NCBI SRA, ENA, GSA)并提供登录号。
生物信息学分析 (Bioinformatic Analysis):
16S rRNA数据处理与微生物群落分析:
原始数据质控: 使用的软件(如FastQC, Trimmomatic, Cutadapt, QIIME2 q2-cutadapt)及其参数(如接头序列、低质量碱基切除阈值、最短读长保留)。
序列拼接与去嵌合体: 双端reads拼接软件(如FLASH, PEAR, QIIME2 q2-vsearch join-pairs)及参数。嵌合体去除软件(如UCHIME, VSEARCH uchime_denovo, DADA2)及参数。
OTU聚类/ASV降噪: OTU聚类软件(如VSEARCH, UPARSE)及相似度阈值(如97%),或ASV生成软件(如DADA2, Deblur)及核心参数。
物种注释: 参考数据库名称及版本(如SILVA v138.1, Greengenes v13_8, GTDB R214),注释方法/软件(如QIIME2 feature-classifier, VSEARCH, BLASTn)及分类阈值(如相似度、置信度)。
Alpha多样性: 计算的指数(如Observed species, Chao1, ACE, Shannon, Simpson, Faith's PD),使用的软件(如QIIME2 q2-diversity, R vegan包)。
Beta多样性: 采用的距离算法(如Bray-Curtis, Jaccard, Unweighted/Weighted UniFrac),排序分析方法(如PCoA, NMDS),使用的软件。
宏基因组数据处理与ARGs分析:
原始数据质控: 同16S rRNA,可能还包括去除宿主基因组污染(如使用Bowtie2比对到参考宿主基因组)。
序列组装 (可选但推荐): 组装软件(如MEGAHIT, IDBA-UD, SPAdes)及其关键参数(如k-mer设置)。组装质量评估(如N50, L50)。
基因预测 (若组装): 基因预测软件(如Prodigal, MetaGeneMark)及其模式(如宏基因组模式)。
ARGs注释: 参考数据库名称及版本(如CARD, SARG, ResFinder),比对工具(如DIAMOND, BLASTP, HMMER),筛选阈值(如E-value, identity, coverage)。
ARGs定量与标准化: 计算方法(如RPKM, TPM, copies per cell, copies per Gb - 如参考论文公式1)。若按细胞当量标准化,需说明单拷贝标记基因的选择和计算方法。
MGEs注释: 参考数据库(如Aclame, ISfinder, INTEGRALL, PlasmidFinder),比对及筛选参数。
VFs注释: 参考数据库(如VFDB),比对及筛选参数。
ARGs宿主菌与可移动性分析: MGEs与ARGs在contigs上的共现或邻近分析标准。宿主菌鉴定方法(如基于contig的物种注释,或将带有ARG的ORF比对到RefSeq,使用MEGAN等工具)。
网络分析: 微生物与ARGs(或其他基因)的共现网络构建。丰度数据类型(相对丰度、有无),相关性算法(如Spearman, Pearson),阈值(相关系数、P值),网络构建和可视化软件(如R igraph/ggraph, Gephi, Cytoscape)。
统计分析 (Statistical Analysis):
结果部分应客观、清晰地展示研究发现,通常按照逻辑顺序组织,并配合高质量的图表。避免在结果部分进行过多的解释和讨论。
放牧对土壤理化性质的影响 (若有测定):
放牧对土壤微生物群落结构的影响 (基于16S rRNA数据):
放牧对土壤ARGs赋存特征的影响 (基于宏基因组数据):
放牧对ARGs横向转移潜能 (HTP) 的影响:
放牧对ARGs潜在致病风险的影响:
ARGs的宿主菌分析与微生物-ARGs关联网络:
多重耐药基因 (MRGs) 风险的重点分析:
图表规范与统计学标注:
讨论部分是对研究结果的深入解读、与前人研究的比较、潜在机制的探讨、研究意义的升华以及对研究局限性和未来展望的陈述。
核心发现总结与解读 (Summary and Interpretation of Key Findings):
与已有研究的比较与联系 (Comparison with Existing Literature):
将本研究的结果与国内外已发表的相关研究进行比较。关注点可以包括:
讨论本研究结果与前人研究一致性的地方,可以增强结论的可靠性。
分析与前人研究不一致的原因,可能涉及研究区域的地理气候差异、土壤类型、放牧强度、牲畜种类、当地抗生素使用情况、研究方法(如测序深度、分析流程)等。
潜在机制探讨 (Elaboration of Potential Mechanisms):
生态学意义与风险启示 (Ecological Implications and Risk Concerns):
研究的创新性与优势 (Strengths and Novelty of the Study):
客观评价本研究的特色和贡献。例如:
研究局限性 (Limitations of the Study):
诚实地指出本研究存在的不足之处,这体现了科学的严谨性。例如:
结论与展望 (Conclusions and Future Perspectives):
用几句精炼的语言概括本研究的核心结论。
基于研究发现和局限性,提出未来值得进一步研究的方向。例如:
 游客
游客

 扫码免费加入loez官方交流QQ群,与各位大佬一起交流技术、讨论问题。
扫码免费加入loez官方交流QQ群,与各位大佬一起交流技术、讨论问题。 